

알림
이 포스팅은 2019년 Harvard STAT115강의를 요약한 것임을 알려드립니다.
다룰 토픽
- Cel file / cdf file
- Array normalization: Loess, Quantile normization
- Normalization quality check : MA plot
- Gene expression index (GEI) : RMA
Affymetrix GeneChip Arrays
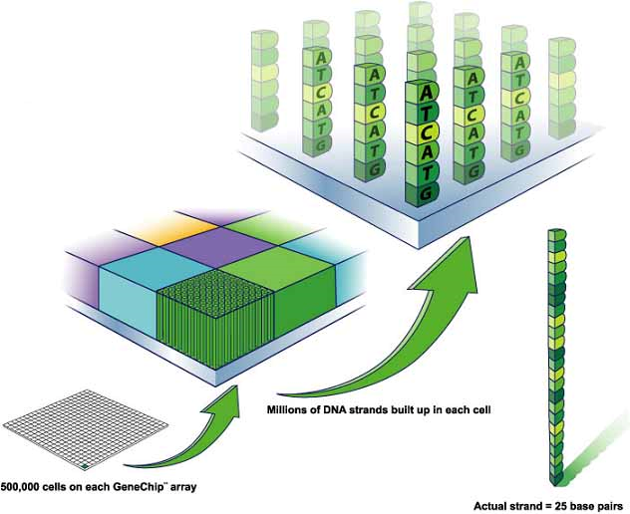
1995년경 Stanford 대학에서 발명한 Gene expression을 측정하는 도구로, 현재 Affymetrix라는 회사서 판매하고 있다. 아래 그림은 microarray의 구성을 나타내고 있다. 각 프로브(칸) 마다 여러 개의 미리 세팅된 염기서열이 있다. 이에 대응되는 (예를 들면 A-T, C-G) 우리가 분석하고자 하는 DNA의 염기서열이 달라붙음으므로 염기서열을 알아낼 수 있다.

Microarray방법은 실험 오차를 줄이기 위하여 최소 3개의 replicates를 두는 것이 좋다. 그러나, 현재, 기술의 발전으로 genome-wide expression array는 잘 쓰이지 않는다. 왜냐하면 대안인 RNA-seq의 비용이 크지 않고, 더 정확한 (실험 오류가 적은)분석을 할 수 있고, 이미 공공데이터가 많이 개방되어있으며, RNA-seq로 여러 data analysis를 할 수 있다는 장점이 있기 때문이다.
- Microarray로 이제 새로운 데이터를 생성하지는 않지만, NCBI에는 GEO (Gene expression omnibus)라고 불리는 여러 microarray로부터 측정된 gene expression level 데이터가 존재한다.
Affymetrix Microarray Data
Microarray로 측정된 데이터는 cell file에 저장된다. cell file의, raw data는 다음과 같이 생겼다.
X Y MEAN STDV NPIXELS 701 523 311.0 76.5 16 702 523 48.0 10.5 16
- (X, Y) : 프로브 위치, 하나의 프로브에는 여러개의 PIXEL이 존재
- MEAN : 평균 gene expression level
- NPIXELS : 각 프로브 마다 가장 중간의 NPIXELS개의 픽샐만을 선택해 expression level을 산출. (경계는 오염될 수 있기 때문.)
Microarray 디자인에 대한 정보는 cdf file에 저장된다.
- (X, Y)에 해당하는 프로브가 어떤 sequence에 해당되고 어떤 transcript를 타겟팅하는지에 대한 정보
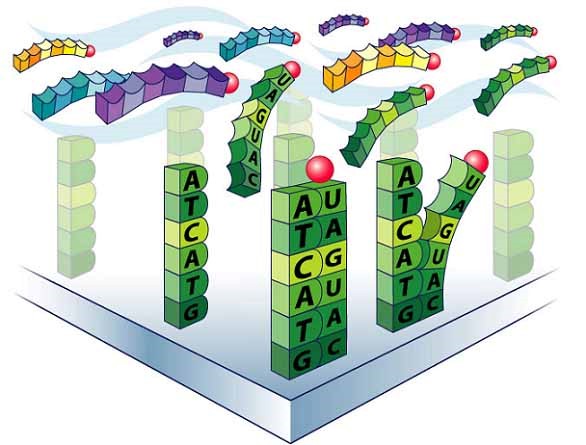
- MM (MisMatch) probes는 거의 항상 PM (Perfect Match) probe 바로 옆에 있으므로 서로 비교할 수 있다.
Perfect Match (PM) and MisMatch (MM) : control for cross hybridization
- 프로브를 생산 (hybridization) 하는 과정에서 RNA를 cDNA로 복제하는데, 이 과정에서 cDNA가 원 RNA와 염기서열이 동일하다면 PM, 동일하지 않다면 MM이라고 한다.
- MM은 분석에서 background noise를 측정하는 데에 사옹된다. PM과 MM의 expression level의 차이를 비교해보 real signal이 무엇이고 noise가 무엇인지 식별하는 데 도움이 된다.
- MM은 보통 PM보다 lower한 gene expression level을 가진다. (항상은 아님, due to cross-hybridization)
- 나중에는 PM만을 사용하게 됨 (후에 설명함).
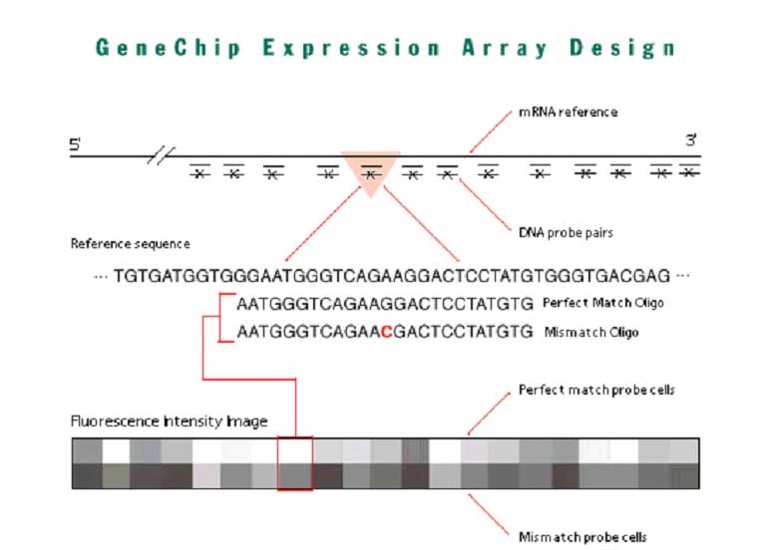
Probe Definition file (cdf file)
- 하나의 gene이더라도 여러 요인에 의해 다른 RNA나 transcript가 생성되기 때문에 데이터는 모두 다르고, cdf file 또한 여러 버전으로 나누어져 있다. (cdf for genes, cdf for transcripts, …)
- 분석의 목적에 따라 cdf 파일을 알맞게 선택해야 한다. 만약 reference gene detection (transcript만 알고 싶어하는 경우)이 목적이라면 그에 해당하는 cdf파일을 선택해야 한다 (refseqmRNA).
- 가장 최신의 cdf annotation 사용하는 것이 좋다. (과거의 annotation과 30%가 다를 수 있음, bioConductor를 최신버전으로 사용)
Microarray Normalization
Microarray분석을 위하여 cdf파일과 cell파일을 받게 되면 가장 먼저 해야할 것은 Normalization이다. 이는 여러 어레이를 사용하여 각각 다른 RNA를 분석할 때, 어레이 간 편차 (다른 스캐너를 사용하거나, 실험 날짜가 다른 이유로 등으로 발생함) 를 줄여주기 위한 것이다. Normalization의 목적은 다음과 같다. Normalization을 하느냐 안하느냐에 따라 실험 결과에 영향을 많이 미치므로 꼭 하는것이 좋다.
- Biological variation은 유지하고, experimental variation은 최소화하여 마이크로어레이간 비교를 가능하게 한다.
- 가정 : 두 샘플(condition)간 genes / probes가 대부분 동일해야 한다.
1) Median Scaling
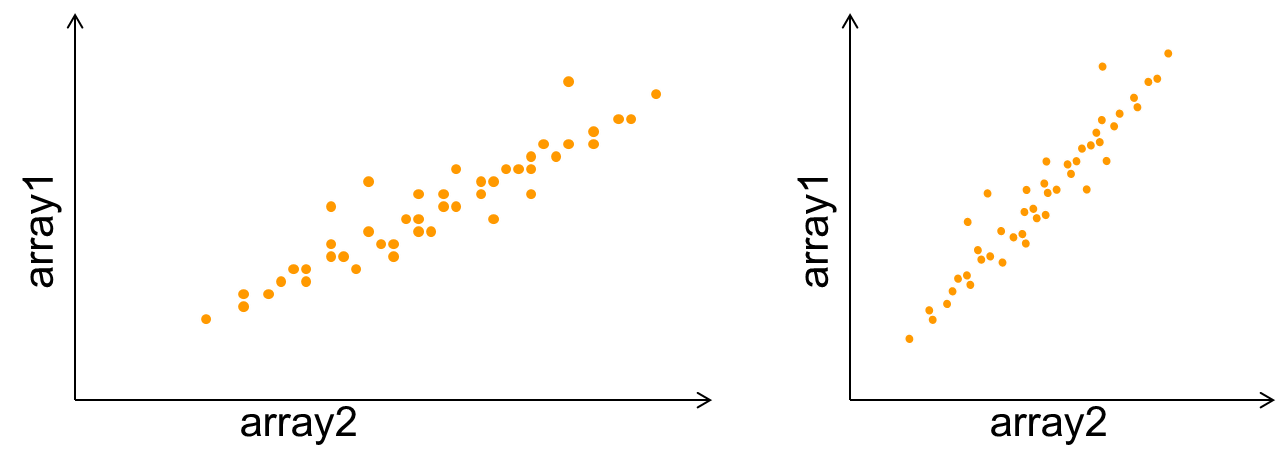
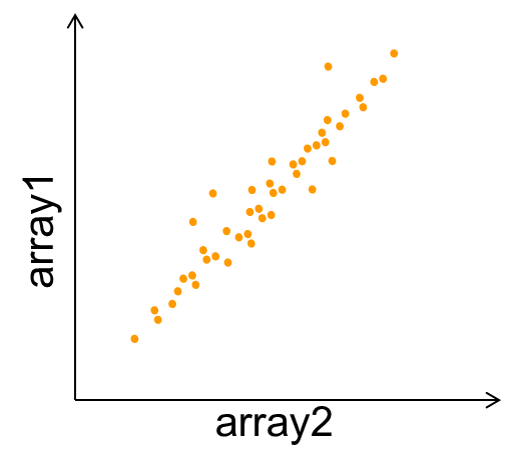
- 그림에서 array1과 array2는 동일한 array를 사용했다고 가정하고 각각 sample1과 sample2를 사용했다고 가정한다. 점은 각 probe의 gene expression level을 나타낸다.
- 두 샘플 간 median이 거의 동일하고 range도 거의 동일하다고 할 때, 다음의 공식으로 Normalization을 할 수 있다: X’ = (X-C1)*C2
2) LOESS (LOcally WEighted Scatterplot Smoothing)
하지만, 아래 왼쪽 그림 같이 두 어레이의 관계가 비선형인 경우도 있다. 즉 하나의 샘플(어레이)에서 시그널이 매우 잘 측정되고 (high gene expression level), 다른 샘플(어레이)에서는 이가 잘 나타나지 않는 경우도 있다.
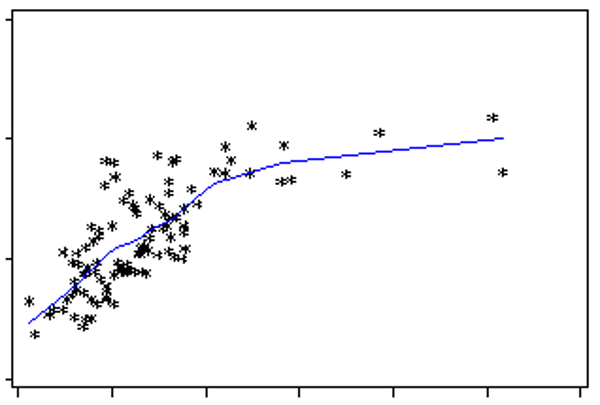
이런 경우 어떻게 Normalization을 수행할 수 있을까? 이럴때 사용하는 것이 LOESS이다. LOESS는 Piecewise linear regression을 사용하여 구간 별로 Normalization을 수행한다. 이러면 이상적인 normalization을 수행할 수 있다. (위 오른쪽 그림)
- 단점 : LOESS normalization을 사용하는 경우 부득이하게 한 array를 reference array로 사용하여 다른 array를 normalization해야 하는데, 이 reference가 되는 array가 좋은 array라는 보장이 없다.
3) Quantile Normalization
LOESS의 reference array의 quality문제를 해결해 줄 수 있는 normalization방법이 Quantile Normalization이다. 이 Quantile normalization은 현재로써는 최적의 normalization 방법으로 여겨지고 있다.
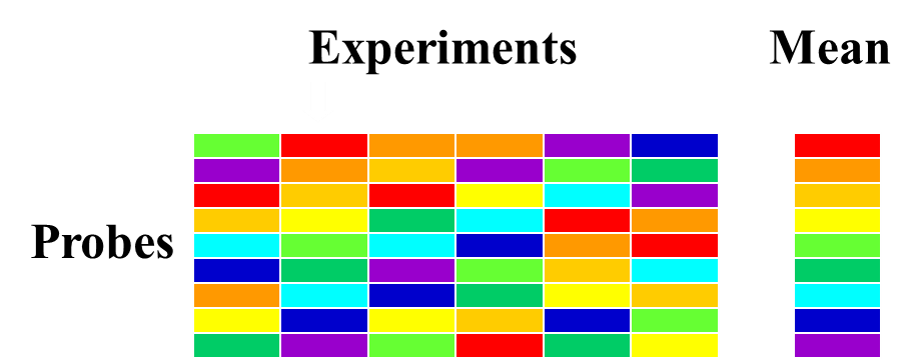
- 가정 : genes / probes의 샘플 간 차이가 거의 없다고 가정한다.
- 각 Experiment의 quantile(색상)마다 평균을 계산하여 각 프로브의 quantile에 대체한다.
- 이 평균값을이 새로운 reference sample역할을 하게 된다.
- 모든 experiment가 똑같은 distribution을 같게되는 장점이 있음.
- 장점 : 새로운 샘플이 들어왔을 때 update하기가 쉬움
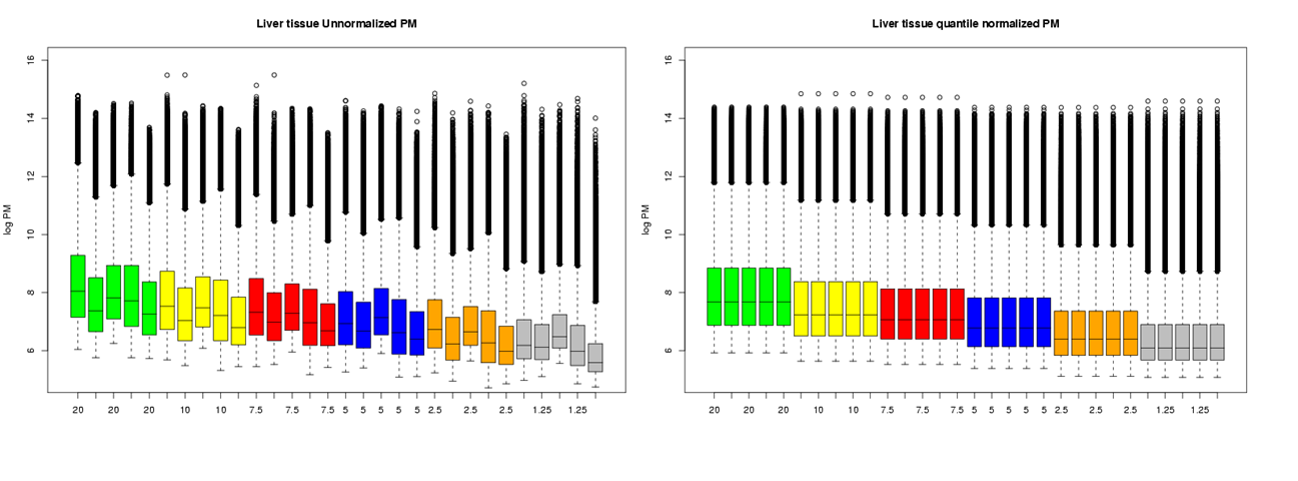
다음 그림은 quantile normalization전과 후의 quantil 별 (색상 별) gene expression level의 boxplot을 보여준다. 그림에서 볼 수 있듯이 quantile normalization이전에는 여러 experimental한 이유 (다른 스캐너를 사용하거나, 기계, 프로세싱 과정)로 각 level이 다름을 알 수 있다. 그러나 quantile normalization후에는 각 quantile별로 sample들이 같은 expression level을 가지는 것을 알 수 있다.
MvA Plot: For Normalization Quality
Normalization이 잘 되었는지를 확인하기 위해서, MvA Plot을 그려볼 수 있다. 만약 샘플 R (Red의 약자, 예전에 빨간색으로 염색을 해서 붙여짐)과 G (Green의 약자)가 있다고 할 때, 각 프로브에 되한 gene expression level의 log값을 그래프로 나타낸 것이 MvA plot이다. (logR vs logG). 그래프가 diagnal하면 normalization이 잘 되었다고 볼 수 있다.
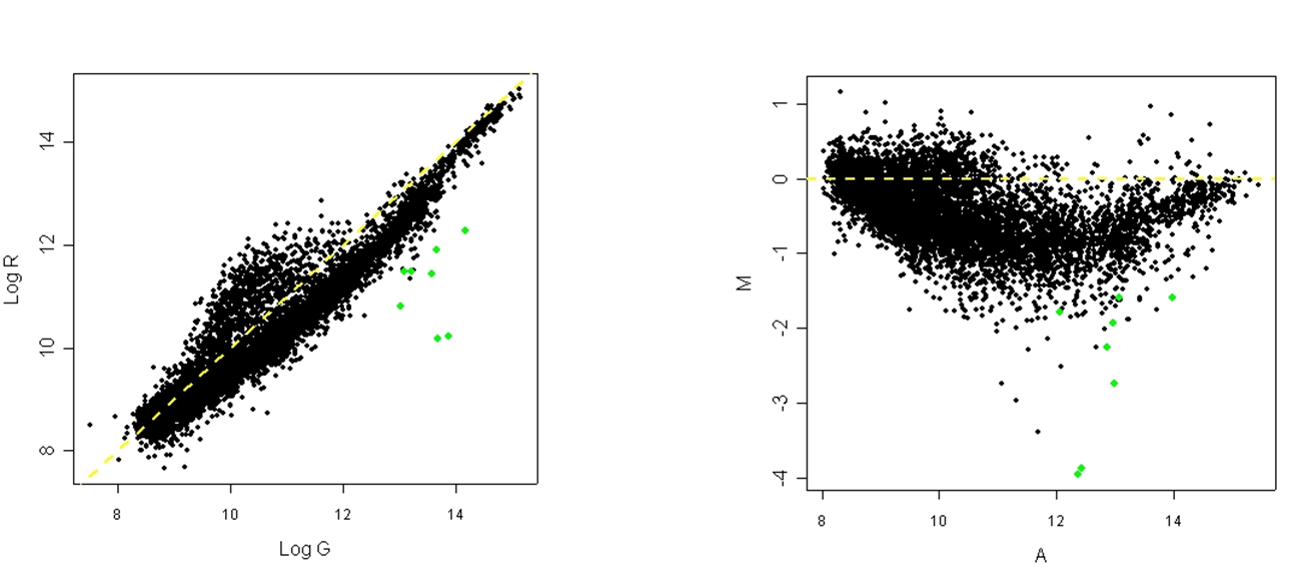
위 왼쪽 그림은 MvA plot의 예시를 보여주는데 각 프로브를 나타내는 점들이 diagonal한 선에 응집되어있지 않으므로 normalization이 잘 되었다고 볼 수 없다.
또 다른 방법으로는 MA Plot (위 오른쪽 그림)을 그리는 것인데, 여기서 M은 M = logR - logG로 log값의 차이를 나타내고, A는 A = (logR + logG) / 2로 기하평균을 나타낸다. MA plot의 경우 점들이 수평선 h=0에 모여있어야 잘 normalization되었다고 할 수 있다.
Gene Expression Index
데이터를 normalize했으면 그 다음은 Gene expression index(GEI)를 살펴봐야 한다. Gene expression index는 얼마나 이 gene이 expressed되었는지를 정량적으로 나타내주는 수치이다. 앞서 다루었듯이 하나의 마이크로어레이에는 여러개의 probe 다발 (probe set, 9 x 9 or 16 x 16 등등)이 존재하며 이 다발하나에 특정 DNA염기서열이 달라붙음으므로써 gene expression level을 측정하게 해 준다. 보통 한 microarray에서 10~20개의 DNA염기서열을 분석할 수 있다.
프로브를 hybridization하는 과정에서 Perfect Match(PM)과 MisMatch(MM)가 발생하고, PM과 MM 주로 양옆에 존재하게 된다. 결과적으로 우리는 10~20쌍의 분석하고자 하는 DNA염기서열에 대해 PM과 MM에 대한 mean gene expression level값을 얻을 수 있다. (아래 그림 참고). 파란색 선은 PM을 나타내고 빨간색 선은 MM을 나타낸다. 그림을 보면 보통 PM이 MM보다 큰 값을 가진다. 하지만, array의 잘못된 설계나, outlier에 의해 MM이 더 높은 경우도 있으며, 이런 상황이 발생되지 않게 해결해주어야 한다.
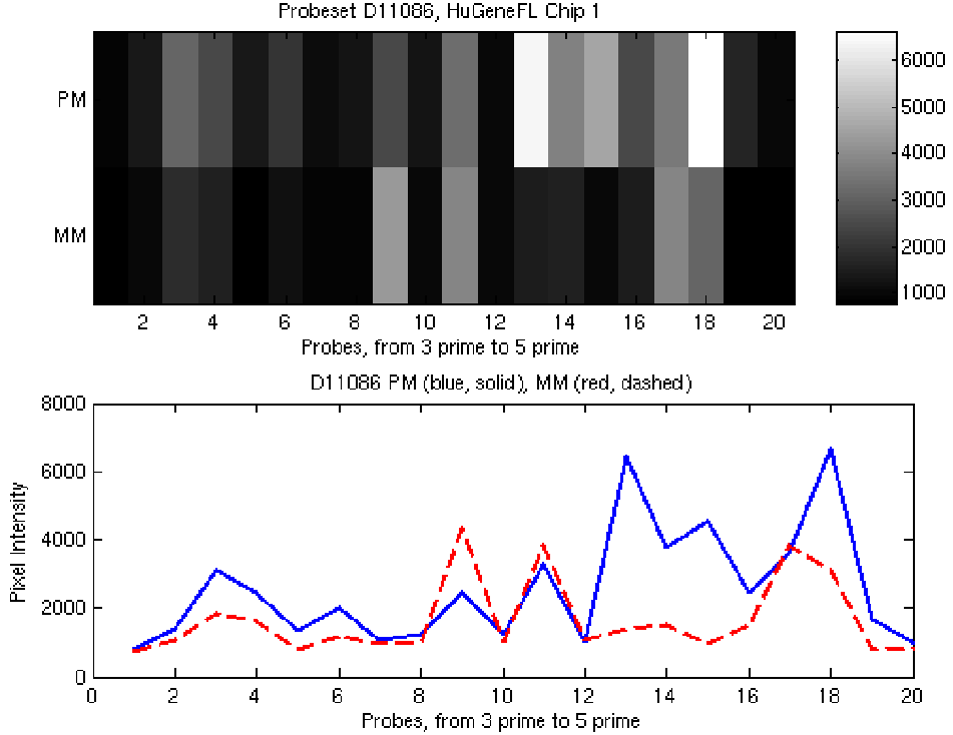
이러한 10~20쌍의 숫자들을 하나의 숫자로 요약하는 것이 GEI (Gene Expression Index)이다.
1) MAS4 : Average difference of PM and MM pair를 사용하는 방법
초기 Affymetrix는 GEI를 다음과 같이 PM과 MM간 차이의 평균을 계산하는 방식으로 산출하였다.
여기서 A는 PM과 MM쌍의 적절한 집합을 말하며, A 는 쌍의 갯수를 의미한다. - 여기서 적절한 집합이란, 최대, 최소값, 평균에서 3*sd 만큼 떨어진 값을 제외한 PM, MM값을 의미한다.
하지만 단순히 Average Difference를 사용하는 것은 너무 naive하고, 30~40%의 프로브가 손실된다는 단점이 있으며, 차이를 계산하는 과정에서 negative value를 가지게 된다 (논리적으로 맞지 않음)는 단점이 있다.
2) RMA (Robust Multi-chip Analysis)
이러한 단점을 해결하기 위해 여러 대안들이 제시되었다. 그 중 가장 유명한 RMA는 Irizarry & Speed, 2003에 의해 알려졌다. RMA는 MM는 전혀 사용하지 않고 PM만으로 Noise와 Signal을 분류한다. 자세한 Step은 다음과 같다.
(Step1) Probe intensity background correction (background subtraction)
- Observed PM 은 Signal과 background noise가 합해진 것으로, Signal은exponential 분포를 따른다고 가정하고, background ~ Normal이라 가정하자. 또한 signal은 항상 양수라고 가정하자. 그러면, 다음과 같은 식이 성립한다.
- 이는 background noise를 추정하기 위해 array에 있는 모든 PM 프로브로부터의 Signal을 활용하는 것이다. 식을 자세히 보면, 모든 PM에 대한 Signall의 기대값이 PM에 대한 전체적인 background noise이라는 것을 알 수 있다.
(Step2) Quantile normalize the log transformed background adjusted PM
- 그 다음은, array간 차이로인해 발생한 signal의 차이, 혹은 RNA의 양이 다름에 따라 발생한 signal강도의 차이를 normalize하는 과정이 필요하다. 이를 위해 log(PM)을 적용시킨다.
- 아래 그림의 왼쪽을 보면 PM의 average signal (x축) vs sd (y축)을 나타낸 것이다. 이 그림을 보면, signal이 작은 probe들은 variation (= noise)도 작은 반면, signal이 probe들은 variation (= noise)도 대체로 큰 것을 알 수 있다. 따라서, 모든 probe signal을 그냥 평균을 내면 high signal을 가진 probe가 크게 영향을 주므로 이 effect를 최소화하기 위하여 log를 취한다.
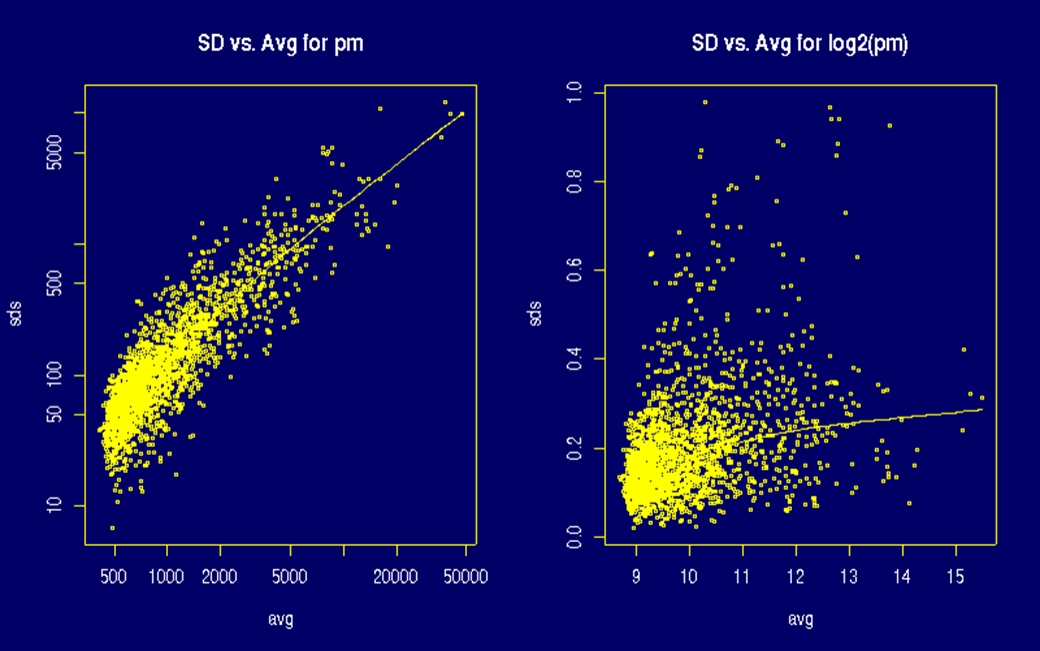
- 오른쪽 그림은 log(average signal) (x축)을 취한 것이다. 그림을 보면, 완벽하지는 않지만 sd (= variation)이 어느정도는 0근처에 모여있어 log를 취하기 전 보다 noise가 줄어들었음을 알 수 있다. 이렇게 log를 취함으로써, 약한 PM signal과 강한 PM signal을 모두 고려하여 GEI를 계산할 수 있다.
(Step3) Robust probe summary
- 수식으로 위의 과정을 설명해보자. i를 sample index, j를 probe index라고 하였을 떄, sample i의 probe j에서의 PM값은 로 나타낼 수 있다. 를 i-th 샘플에서의 transcript의 concentration이라고 하고, 를 j-th probe의 probe effect라고 하면, 로 나타낼 수 있다. (생물학적 사실). Normalization을 위해 Step 2와 같이 PM에 로그를 취하면 다음과 같다.
- 이를 다르게 나타내면 다음과 같다.
- 여기서 는 array by array로 background subtraction을 하는 것이고, 는 n개의 샘플에 대한 quantile normalization후에 log를 취하는 것을 뜻한다. 이 값을 이용하여 와 를 추정한다. 여기서, 는 expression index를, 는 probe effect, 는 최종 noise를 나타낸다.
- 와 를 추정은 Median polish (알아보기!) 를 통해 반복적으로 와 를 적합시켜서 구한다. 최종적으로 expression index ()를 구할 수 있다.
- RMA expression index는 로그스케일인것에 주목하자.
- 이 방법은 빠르며, outlier에 대해 robust하다는 장점이 있다.
- RMA방법이 개발됨에 따라 Affymetrix는 더이상 MM probe를 디자인하지 않고 있다.
Method Comparison
RMA등 새로운 GEI를 계산하기 위한 방법들이 고안되었을 때, 어떻게 성능을 평가할까에 대한 물음이 있을 수 있다. 여러 methods를 비교하기 위해 Spike-in RNA라는 실험이 고안되었다. 동일한 RNA가 있다고 가정할 때, 이 RNA를 절반으로 나눈다 (샘플 1, 샘플 2). 그런 후, 한쪽 절반 (sample 1)에는 다른 RNA를 추가하여 섞는다. (아래 그림에서는 1~11번까지 RNA가 추가 되었다.)
- 왼쪽 그림은 MAS4를 사용하였고, 오른쪽 그림은 RMA를 사용하여 MA plot을 그린 것이다. x축은 gene의 average expression level을, y축은 두 샘플 (샘플 1, 샘플 2)에서 측정한 expression level의 차이이다. 각 point는 하나의 gene을 나타낸다.
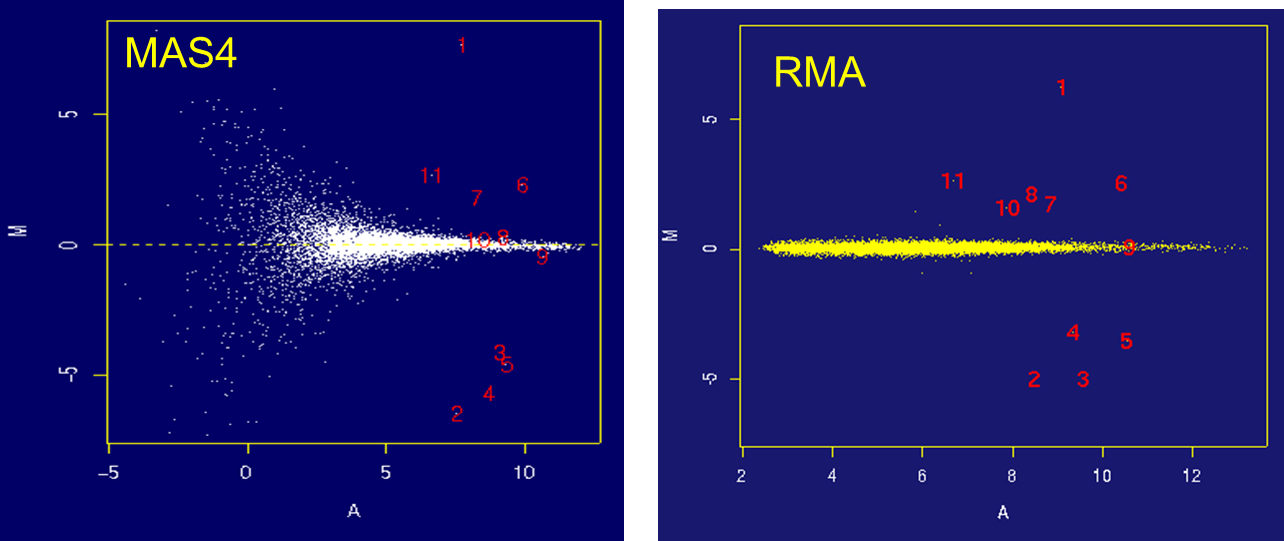
- 샘플 1과 샘플 2는 11개의 추가된 RNA를 제외하고는 동일하므로, expression level의 차이가 11개의 점 이외에는 없어야 한다 (즉 y = 0). 하지만 MAS4를 보면 low average expression level (x축의 왼쪽편)에서 많은 noise가 관찰된다. 반면 RMA는 두 샘플간의 차이가 거의 없고, 또한 추가된 11개의 gene에 대해서는 차이를 명확히 나타내고 있어 좋은 성능을 내는 것을 알 수 있다.
- x축을 보면 MAS4는 범위가 -5 ~ 10 이지만 RMA는 log scale을 사용하므로 범위가 2 - 12임을 알 수 있다.
최종 데이터셋
결과적으로 RMA gene expression level을 계산한 후 우리는 각 행이 gene 혹은 transcripts로 이루어지고 (cdf파일에 따라 다름), column은 각 샘플 혹은 condition으로 이루어진 큰 matrix를 만들 수 있다. 이 matrix를 분석에서 활용한다.


